

The mutations that drive cancer formation are often found in “hub” genes that regulate many aspects of cell growth and survival. But these key genes are not always good therapeutic targets — some are even considered “undruggable.” In the latest issue of GENETICS, Bailey et al. identify a strategy for fighting cancer cells that carry a mutation in one such difficult-to-target hub gene, FBW7. This tumor suppressor is mutated in approximately 6% of all cancer cases, including around a third of cases of T-cell acute lymphocytic leukemia and cholangiocarcinoma.
The FBW7 protein is part of the SCF ubiquitin ligase complex that marks other proteins for selective degradation. Many of these substrates are involved in oncogenesis (cancer formation), including the cell cycle regulator cyclin-E and the cell death inhibitor MCL1. Without FBW7 keeping them in check, these cancer-promoting proteins accumulate in the cell and can trigger uncontrolled division.
Even though FBW7’s tumor suppressor function is well understood, little is known about how the mutant cells might be targeted for treatment. That’s because inactivating the function of a tumor suppressor gene (e.g. with a drug) would encourage, not inhibit cancer, while many of FBW7’s oncogenic substrates are themselves difficult to manipulate with pharmaceuticals.
The authors approached this problem by screening for “synthetic lethal” partners of FBW7, which are proteins needed for cell survival when FBW7 is non-functional. They screened approximately 16,000 human genes by knocking down their expression in fbw7 mutant colorectal cancer cells and FBW7 wild-type cells, looking for proliferation effects that were specific to the mutants.
One of the candidates identified in the screen was BUBR1, which encodes a component of the mitotic spindle assembly checkpoint (SAC). This checkpoint prevents cell division from proceeding until all chromosomes are correctly attached to the spindle apparatus. When BUBR1 expression is knocked down in fbw7 mutant cells, the cells proliferate slower than wild-type and become more prone to losing and gaining chromosomes through cell division errors. This vulnerability of fbw7 cells was confirmed by tests with other SAC components.
Why do fbw7 cells have such a critical need for spindle assembly surveillance? One part of the answer is that the cells have dysregulated cyclin E. This is suggested by the fact that dampening expression of cyclin E rescues fbw7 mutant cells from their dependence on BUBR1. Cyclin E is not the whole story, however, because boosting levels of this protein alone was not enough to make FBW7 wild-type cells sensitive to SAC loss; this was only achieved by overexpressing another FBW7 substrate, MCL1, along with a form of cyclin E sometimes seen in cancer cells.
The results show that in this cell culture model, fbw7 mutant cells depend on the SAC for their cancerous potential, likely because they have cell cycle defects that necessitate extra time for spindle assembly. Without this breathing room, the dividing cells may be more prone to lethal chromosome segregation mistakes.
The authors suggest that other cancer cell types — many of which experience chronic chromosome instability — might also depend on the SAC. They suggest that exploiting this weak spot could be a promising strategy for developing anticancer drugs, especially since blocking SAC function could have fewer side-effects than existing anti-mitotic drugs. Though there is a long way to go before this idea could be applied in the clinic, the “undruggable” target FBW7 may have pointed the way to a more accessible chink in cancer’s armor.
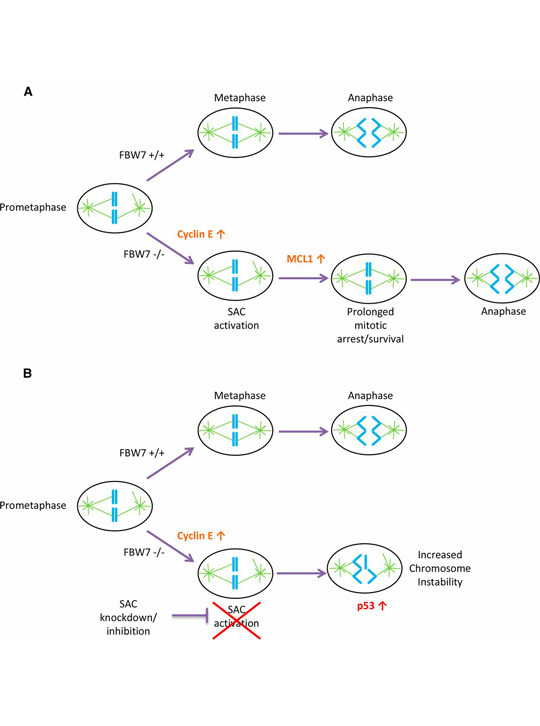
A model for SAC dependence in cells lacking FBW7. From Baikey et al. (A) FBW7 +/+ cells in prometaphase efficiently align their chromosomes and correctly segregate properly in anaphase with less reliance on the SAC. FBW7 −/− cells have an increase in cyclin E, which causes problems in mitosis and may lead to SAC activation. FBW7 −/− cells can survive prolonged SAC activation in part because of stabilization of MCL1 allowing more time for chromosome alignment. (B) A decrease in SAC activation by knockdown of BUBR1 may significantly shorten the time available for chromosome alignment in FBW7 −/− cells, resulting in improper chromosome segregation and intolerable levels of chromosome instability.
Citation:
Bailey, M. L., Singh, T., Mero, P., Moffat, J., & Hieter, P. (2015). Dependence of Human Colorectal Cells Lacking the FBW7 Tumor Suppressor on the Spindle Assembly Checkpoint. Genetics, 201(3), 885-895. Doi: 10.1534/genetics.115.180653
Cristy Gelling is a science writer, lapsed yeast geneticist, and former Communications Director at the GSA.
View all posts by Cristy Gelling »Read more in
-

Thank you, GSA community!
Thank you for being a member of the Genetics Society of America! As GSA’s current president, I am writing to tell you about Society projects and initiatives that we hope you will find useful in advancing your science and your career. Scientific research is a collaborative and exciting endeavor. Scientific societies like GSA exist to…
-

Where are they now? Rosalind Franklin Young Investigator Award recipients share updates on their research
Rosalind Franklin Young Investigator Award applications are open–make sure you submit your application or nomination of a colleague by September 30, 2024.
-

University of Minnesota researchers map genome of the last living wild horse species
The study, published in G3: Genes|Genomes|Genetics, is part of larger conservation efforts to save Przewalski’s horse.
-

Congratulations to the Spring 2024 DeLill Nasser Awardees!
GSA is pleased to announce the recipients of the DeLill Nasser Award for Professional Development in Genetics for Spring 2024! Given twice a year to graduate students and postdoctoral researchers, DeLill Nasser Awards support attendance at meetings and laboratory courses. The award is named in honor of DeLill Nasser, a long-time GSA supporter and National Science Foundation…
-

Carolyn Damilola: an NFS Rising Scientist on a lifelong quest to learn more
Carolyn Damilola is an NFS Rising Scientist from Nigeria doing respiratory system research and paving the way for scientists from underrepresented communities through mentorship.
-

What does a good microgrant proposal look like?
Members of the Microgrant Review Committee share their tips for a successful proposal.
-

The first piece of the facial recognition puzzle
New research in GENETICS gives a first peek at the molecular pathway involved in recognizing faces.
-

New Senior Editor Amy MacQueen joins GENETICS
A new senior editor is joining GENETICS in the Genome Integrity and Transmission section. We’re excited to welcome Amy MacQueen to the editorial team.
-

Block party on the zebrafish sex chromosome
Research in G3 identifies a gene regulatory block of the zebrafish genome responsible for overseeing the maternal-to-zygotic-transition.
-

Unraveling the mysteries of duckweed: epigenetic insights from Spirodela polyrhiza
Research published in G3 offers insight into the impact of DNA methylation on clonal propagation in asexually reproducing plants.
-

A microbiologist’s quest to understand CRISPR in bacterial self-defense
2024 Genetics Society of America Medal recipient Luciano Marraffini determined how CRISPR-Cas systems destroy genetic targets with precision, paving the way for gene editing technology development.
-

Unlocking mysteries of trait and disease heritability in dogs
2024 Edward Novitski Prize recipient Elaine Ostrander, a pioneer of the domestic dog model, discovered numerous genes affecting dog size, morphology, behavior, and disease susceptibility—many of which have relevance in humans.
-

GSA and collaborators Personal Genetics Education & Dialogue and Reclaiming STEM Institute launch NSF-funded BIO-LEAPS project to support culture change in genetics
We are thrilled to announce that the Genetics Society of America (GSA) is collaborating with the Personal Genetics Education & Dialogue (PGED) based in the Department of Genetics at Harvard Medical School, and the Reclaiming STEM Institute (RSI) on a Leading Culture Change Through Professional Societies of Biology (BIO-LEAPS) grant from the U.S. National Science…
-

Daman Saluja: Navigating Science and Policy in India
In the Paths to Science Policy series, we talk to individuals who have a passion for science policy and are active in advocacy through their various roles and careers. The series aims to inform and guide early career scientists interested in science policy. This series is brought to you by the GSA Early Career Scientist…
-

A fly geneticist’s journey into discovering rules of organ development
2024 George W. Beadle Award recipient Deborah Andrew discovered new genes and pathways in Drosophila salivary gland organogenesis. Now, her work can help optimize cell secretion in therapeutic applications and fight malaria.
-

Małgorzata Gazda: How receiving the DeLill Nasser Award helped her land her dream job
Have you ever experienced an event that changes the course of your life, or in this case, your career? Małgorzata (Gosia) Gazda is Assistant Professor at the University of Montreal and in 2022, she received the DeLill Nasser Award for Professional Development in Genetics, which she used to attend and present at the 2022 Population,…
-

Hongyu Zhao joins GENETICS as new Senior Editor
A new senior editor is joining GENETICS in the Statistical Genetics and Genomics section. We’re excited to welcome Hongyu Zhao to the editorial team.
-

GSA Member Julio Molina Pineda Receives DeLill Nasser Award, Shines at TAGC 2024
“At any career stage, the GSA membership is an amazing investment for any genetics professional!” Julio Molina Pineda is a PhD Candidate in Cell and Molecular Biology and a Research Assistant at the University of Arkansas, and a Doctoral Academy Fellow at the Lewis Lab. In 2023, Julio was awarded the DeLill Nasser Award for…
-

In Memoriam: Ellsworth Herman Grell (1932–2023), a pioneer of Drosophila genome engineering and annotation
Ellsworth (Ed) Grell blessed the Drosophila community through three enduring legacies: as a pioneer of chromosome mechanics, as a primary organizer and synthesizer of genetic knowledge in Drosophila, and as a graceful mentor to those fortunate to have known him personally. Ed grew up in rural Nebraska, completed his undergraduate studies at Iowa State, and…
-

Congratulations to the #Fungal24 Poster Award winners!
We are pleased to announce the recipients of the GSA Poster Awards for posters presented at the 32nd Fungal Genetics Conference! Undergraduate and graduate student members of GSA were eligible for the awards, and a hard-working team of judges made the determinations. Congratulations to all! Felicia Ebot Ojong, The University of Georgia My research is focused…
-

Poster presentation tips for TAGC 2024
You’ve been selected to present a poster at The Allied Genetics Conference 2024 in March—you’ve celebrated, made plans to attend, now what? This is an exciting opportunity to showcase your research and engage with fellow members of the genetics community, so you want to make sure you’re prepared. We wanted to offer you some tips…
-

Maximize your TAGC 2024 experience
A guide to all that National Harbor & DC have to offer Are you joining us for The Allied Genetics Conference 2024 in March? Make the most of your #TAGC24 experience in National Harbor! We know the science will keep you busy, but you deserve to unwind and have some fun, so we’ve curated a…
-

Early Career Leadership Spotlight: Sarah Petrosky
We’re taking time to get to know the members of the GSA’s Early Career Scientist Committees. Join us to learn more about our early career scientist advocates. Sarah PetroskyMultimedia SubcommitteeUniversity of Pittsburgh Research Interest I am interested in understanding adaptation that has been happening recently in populations by dissecting the ways that genes underlying an adaptation…
-

TAGC 2024 Early Career Award Winners
GSA is pleased to announce the winners of the early career awards presented at The Allied Genetics Conference 2024. These awards are specific to particular TAGC communities and recognize early career scientists’ outstanding work on their respective research organisms. The awardees will present their talks in keynote sessions at TAGC 2024. Don’t miss the opportunity…
-

Preeminent geneticists recognized with revamped GSA Awards
In 2022, GSA’s Board of Directors launched an audit to review the five major awards conferred by the Society. Today, we are thrilled to announce the recipients of the reimagined GSA Awards, including the new Genetics Society of America Early Career Medal. The scientists honored this year are recognized by their peers for their outstanding…
-

Fly Board funds outreach programs to spread the word about Drosophila research
In 2020, the Fly Board voted to use part of its reserve fund to support efforts to increase trainee participation as well as equity and diversity in the Drosophila community. An awards committee decides how the money will be spent each year, and from 2020–2022, the committee posted a very broad call for applications from…
-

New members of the GSA Board of Directors: 2024–2026
We are pleased to announce the election of four new leaders to the GSA Board of Directors: 2024 Vice President/2025 President Brenda Andrews Professor, University of Toronto It’s an honor to continue my association with the Society by serving as Vice President of the Board of Directors. I have broad knowledge of the ongoing activities…
-

Early Career Leadership Spotlight: Sara McPherson
Sara McPherson Career Development SubcommitteeQueen’s University What is your research interest? What organism do you work with, and what do you want to do in the future? My research interests involve the genetic underpinnings of disease and how understanding the genetics behind key biological processes can be leveraged to develop therapeutics for a variety of…
-

Congratulations to the 2026 Yeast Poster Award recipients!
This year, GSA recognizes outstanding student research at the Yeast Genetics Meeting, honoring exceptional presentations by undergraduate and graduate student members. Award recipients are selected based on both the scientific merit of their research and the clarity of their presentation. Please join us in congratulating this year’s awardees. Nasima AkhterUniversity of Rochester I study the…
-

Tips for finding a scientific narrative
How many robotic talks and lectures have you experienced in your scientific journey? Probably more than you can count! As scientists, we typically prioritize accuracy when communicating our work but sometimes neglect to ensure the audience remains engaged. One way to hold your audience’s attention during presentations is to use a universal communication tool: storytelling. …