

In this month’s editorial, the Editors of GENETICS invite submissions of human genetics research articles. To kick off the journal’s call for papers, the October issue features an article by Brooks and Wall et al. identifying the cause of a single-family disorder and a commentary by Phil Hieter and Kim Boycott on the power of model organisms for understanding rare disease pathogenesis. If you’re at the 2014 ASHG meeting, stop by the GSA booth and pick up a copy!
Susan Brooks had reached a dead end.
Brooks, a medical geneticist at Rutgers Robert Wood Johnson Medical School, was trying to diagnose a boy who had a genetic disorder she had never encountered before. The young patient showed delayed development, microcephaly, recurrent febrile illnesses, seizures, overall slow growth, and several minor craniofacial and limb malformations.
She strongly suspected an X-linked mutation, after finding that two of the boy’s maternal uncles had similar unexplained neurodevelopmental disorders. In addition, the boy’s mother and maternal grandmother displayed non-random patterns of X-inactivation, a clear sign that one of their X-chromosomes carried a harmful mutation.
But the results of a clinical sequencing panel for X-linked intellectual disorders did not fill Brooks with confidence. Of the 81 genes in the panel, the only mutation carried by the boy that was not present in healthy populations was in RPL10, which encodes a component of the large ribosomal subunit. RPL10 was included in the panel because other variants had been implicated in two cases of familial autism, but those cases were nothing like this family’s disorder. Mutations in other ribosomal protein genes can cause Diamond-Blackfan anemia, but this family showed no signs of being anemic.
It was not even clear whether the new variant—a missense mutation—was deleterious to Rpl10 function, as the bioinformatic predictions were ambiguous.
All in all, Brooks needed more proof that the RPL10 mutation was causative. Unless someone stumbled across another family with the same symptoms, this tantalising clue had turned into a dead end.
Then, in 2012, she attended a meeting of the Human Genetics Association of New Jersey, where the invited speaker was Erica Davis from the Center for Human Disease Modeling at Duke University.
The bottleneck in disease genetics, Davis told the audience, is functional annotation. Clinical sequence is accumulating at a rapid rate, but often provides only ‘variants of unknown significance.’ For sequence information to be useful to patients, clinicians need systems for efficiently testing relevance of gene function to disease phenotypes and analyzing the pathogenic potential of mutations. That’s where model organisms come in, said Davis. She
and her colleagues specialize in using zebrafish to provide crucial functional information on variants associated with rare diseases, even those that occur in just one pedigree. One-of-a-kind families, she calls them.
Brooks approached Davis after the talk with the story of the family with the ambiguous mutation. Davis agreed to help. It was just the right task for her new sophomore student, Alissa Wall, who would need a well-defined project for learning the ropes of zebrafish work.
Remarkably, when Wall knocked down the zebrafish RPL10 ortholog, the fish had normal body length but significantly smaller head size. Expression of human RPL10 rescued the phenotype, but human RPL10 bearing the clinical mutation did not. In short, the RPL10 variant caused the zebrafish to develop microcephaly. Not only did Wall’s research generate data for an award-winning honors thesis, it also provided good evidence that it is a loss-of-function mutation and is likely to be responsible for the family’s genetic disorder.
Important as this evidence is, it’s far from a “cure” and has no obvious implications for medical management of the disorder. But it’s a start, and the group has already made progress in understanding how loss of Rpl10 function causes small head size in fish. The lab across the hall from Davis (led by
Christopher Nicchitta) studies mRNA translation, and they helped show that RPL10 knockdown decreases bulk translation in the head and increases apoptosis in the brain. Davis and Nicchitta are pursuing these leads further, hoping to turn up something clinically helpful. For example, they are interested in whether the growth phenotypes are caused by the overall decrease in translation in the brain, or whether the critical trigger is deficiency of a particular subset of proteins.
This research also revealed a new class of ribosomopathy. Now, other components of the large ribosome subunit may be added to the list of candidate genes for rare causes of microcephaly. Thanks to a diverse partnership — patient families, clinical geneticists, zebrafish researchers, and biochemists — Brooks’ dead end is turning into a new beginning.
Read this month’s GENETICS editorial calling for human genetics article submissions.
Read Phil Hieter and Kim Boycott’s commentary on model organisms in rare disease genetic
research.
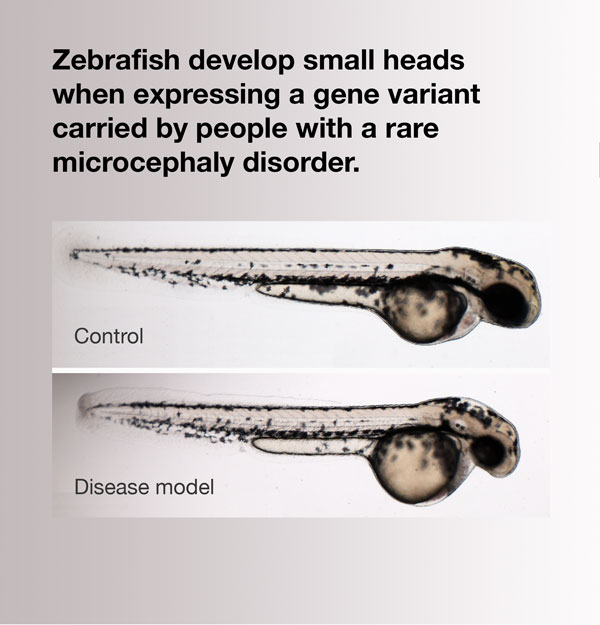
When the gene mutated in patients with a rare disorder is knocked down in zebrafish, the animals develop smaller heads, which is one of the major symptoms of the human disease. Image shows a control zebrafish larva (top) and one in which expression of rpl10 was suppressed (bottom), resulting in normal body length but a proportionately smaller head. From Brooks and Wall et al.
———
Citations:
Brooks S.S., C. Golzio, D. W. Reid, A. Kondyles, J. R. Willer, C. Botti, C. V. Nicchitta, N. Katsanis & E. E. Davis (2014). A Novel Ribosomopathy Caused by Dysfunction of RPL10 Disrupts Neurodevelopment and Causes X-Linked Microcephaly in Humans, Genetics, 198 (2) 723-733. DOI: http://dx.doi.org/10.1534/genetics.114.168211 http://www.genetics.org/content/198/2/723.full
Hieter P. (2014). Understanding Rare Disease Pathogenesis: A Grand Challenge for Model Organisms, Genetics, 198 (2) 443-445. DOI: http://dx.doi.org/10.1534/genetics.114.170217
http://www.genetics.org/content/198/2/443.full
Johnston M. (2014). Humans as a Model Organism: The Time Is Now, Genetics, 198 (2) 441-441. DOI: http://dx.doi.org/10.1534/genetics.114.170225
http://www.genetics.org/content/198/2/441.full
Cristy Gelling is a science writer, lapsed yeast geneticist, and former Communications Director at the GSA.
View all posts by Cristy Gelling »Read more in
-

Thank you, GSA community!
Thank you for being a member of the Genetics Society of America! As GSA’s current president, I am writing to tell you about Society projects and initiatives that we hope you will find useful in advancing your science and your career. Scientific research is a collaborative and exciting endeavor. Scientific societies like GSA exist to…
-

Where are they now? Rosalind Franklin Young Investigator Award recipients share updates on their research
Rosalind Franklin Young Investigator Award applications are open–make sure you submit your application or nomination of a colleague by September 30, 2024.
-

University of Minnesota researchers map genome of the last living wild horse species
The study, published in G3: Genes|Genomes|Genetics, is part of larger conservation efforts to save Przewalski’s horse.
-

Congratulations to the Spring 2024 DeLill Nasser Awardees!
GSA is pleased to announce the recipients of the DeLill Nasser Award for Professional Development in Genetics for Spring 2024! Given twice a year to graduate students and postdoctoral researchers, DeLill Nasser Awards support attendance at meetings and laboratory courses. The award is named in honor of DeLill Nasser, a long-time GSA supporter and National Science Foundation…
-

Carolyn Damilola: an NFS Rising Scientist on a lifelong quest to learn more
Carolyn Damilola is an NFS Rising Scientist from Nigeria doing respiratory system research and paving the way for scientists from underrepresented communities through mentorship.
-

What does a good microgrant proposal look like?
Members of the Microgrant Review Committee share their tips for a successful proposal.
-

The first piece of the facial recognition puzzle
New research in GENETICS gives a first peek at the molecular pathway involved in recognizing faces.
-

New Senior Editor Amy MacQueen joins GENETICS
A new senior editor is joining GENETICS in the Genome Integrity and Transmission section. We’re excited to welcome Amy MacQueen to the editorial team.
-

Block party on the zebrafish sex chromosome
Research in G3 identifies a gene regulatory block of the zebrafish genome responsible for overseeing the maternal-to-zygotic-transition.
-

Unraveling the mysteries of duckweed: epigenetic insights from Spirodela polyrhiza
Research published in G3 offers insight into the impact of DNA methylation on clonal propagation in asexually reproducing plants.
-

A microbiologist’s quest to understand CRISPR in bacterial self-defense
2024 Genetics Society of America Medal recipient Luciano Marraffini determined how CRISPR-Cas systems destroy genetic targets with precision, paving the way for gene editing technology development.
-

Unlocking mysteries of trait and disease heritability in dogs
2024 Edward Novitski Prize recipient Elaine Ostrander, a pioneer of the domestic dog model, discovered numerous genes affecting dog size, morphology, behavior, and disease susceptibility—many of which have relevance in humans.
-

GSA and collaborators Personal Genetics Education & Dialogue and Reclaiming STEM Institute launch NSF-funded BIO-LEAPS project to support culture change in genetics
We are thrilled to announce that the Genetics Society of America (GSA) is collaborating with the Personal Genetics Education & Dialogue (PGED) based in the Department of Genetics at Harvard Medical School, and the Reclaiming STEM Institute (RSI) on a Leading Culture Change Through Professional Societies of Biology (BIO-LEAPS) grant from the U.S. National Science…
-

Daman Saluja: Navigating Science and Policy in India
In the Paths to Science Policy series, we talk to individuals who have a passion for science policy and are active in advocacy through their various roles and careers. The series aims to inform and guide early career scientists interested in science policy. This series is brought to you by the GSA Early Career Scientist…
-

A fly geneticist’s journey into discovering rules of organ development
2024 George W. Beadle Award recipient Deborah Andrew discovered new genes and pathways in Drosophila salivary gland organogenesis. Now, her work can help optimize cell secretion in therapeutic applications and fight malaria.
-

Małgorzata Gazda: How receiving the DeLill Nasser Award helped her land her dream job
Have you ever experienced an event that changes the course of your life, or in this case, your career? Małgorzata (Gosia) Gazda is Assistant Professor at the University of Montreal and in 2022, she received the DeLill Nasser Award for Professional Development in Genetics, which she used to attend and present at the 2022 Population,…
-

Hongyu Zhao joins GENETICS as new Senior Editor
A new senior editor is joining GENETICS in the Statistical Genetics and Genomics section. We’re excited to welcome Hongyu Zhao to the editorial team.
-

GSA Member Julio Molina Pineda Receives DeLill Nasser Award, Shines at TAGC 2024
“At any career stage, the GSA membership is an amazing investment for any genetics professional!” Julio Molina Pineda is a PhD Candidate in Cell and Molecular Biology and a Research Assistant at the University of Arkansas, and a Doctoral Academy Fellow at the Lewis Lab. In 2023, Julio was awarded the DeLill Nasser Award for…
-

In Memoriam: Ellsworth Herman Grell (1932–2023), a pioneer of Drosophila genome engineering and annotation
Ellsworth (Ed) Grell blessed the Drosophila community through three enduring legacies: as a pioneer of chromosome mechanics, as a primary organizer and synthesizer of genetic knowledge in Drosophila, and as a graceful mentor to those fortunate to have known him personally. Ed grew up in rural Nebraska, completed his undergraduate studies at Iowa State, and…
-

Congratulations to the #Fungal24 Poster Award winners!
We are pleased to announce the recipients of the GSA Poster Awards for posters presented at the 32nd Fungal Genetics Conference! Undergraduate and graduate student members of GSA were eligible for the awards, and a hard-working team of judges made the determinations. Congratulations to all! Felicia Ebot Ojong, The University of Georgia My research is focused…
-

Poster presentation tips for TAGC 2024
You’ve been selected to present a poster at The Allied Genetics Conference 2024 in March—you’ve celebrated, made plans to attend, now what? This is an exciting opportunity to showcase your research and engage with fellow members of the genetics community, so you want to make sure you’re prepared. We wanted to offer you some tips…
-

Maximize your TAGC 2024 experience
A guide to all that National Harbor & DC have to offer Are you joining us for The Allied Genetics Conference 2024 in March? Make the most of your #TAGC24 experience in National Harbor! We know the science will keep you busy, but you deserve to unwind and have some fun, so we’ve curated a…
-

Early Career Leadership Spotlight: Sarah Petrosky
We’re taking time to get to know the members of the GSA’s Early Career Scientist Committees. Join us to learn more about our early career scientist advocates. Sarah PetroskyMultimedia SubcommitteeUniversity of Pittsburgh Research Interest I am interested in understanding adaptation that has been happening recently in populations by dissecting the ways that genes underlying an adaptation…
-

TAGC 2024 Early Career Award Winners
GSA is pleased to announce the winners of the early career awards presented at The Allied Genetics Conference 2024. These awards are specific to particular TAGC communities and recognize early career scientists’ outstanding work on their respective research organisms. The awardees will present their talks in keynote sessions at TAGC 2024. Don’t miss the opportunity…
-

Preeminent geneticists recognized with revamped GSA Awards
In 2022, GSA’s Board of Directors launched an audit to review the five major awards conferred by the Society. Today, we are thrilled to announce the recipients of the reimagined GSA Awards, including the new Genetics Society of America Early Career Medal. The scientists honored this year are recognized by their peers for their outstanding…
-

Fly Board funds outreach programs to spread the word about Drosophila research
In 2020, the Fly Board voted to use part of its reserve fund to support efforts to increase trainee participation as well as equity and diversity in the Drosophila community. An awards committee decides how the money will be spent each year, and from 2020–2022, the committee posted a very broad call for applications from…
-

New members of the GSA Board of Directors: 2024–2026
We are pleased to announce the election of four new leaders to the GSA Board of Directors: 2024 Vice President/2025 President Brenda Andrews Professor, University of Toronto It’s an honor to continue my association with the Society by serving as Vice President of the Board of Directors. I have broad knowledge of the ongoing activities…
-

Early Career Leadership Spotlight: Sara McPherson
Sara McPherson Career Development SubcommitteeQueen’s University What is your research interest? What organism do you work with, and what do you want to do in the future? My research interests involve the genetic underpinnings of disease and how understanding the genetics behind key biological processes can be leveraged to develop therapeutics for a variety of…
-

Congratulations to the 2026 Yeast Poster Award recipients!
This year, GSA recognizes outstanding student research at the Yeast Genetics Meeting, honoring exceptional presentations by undergraduate and graduate student members. Award recipients are selected based on both the scientific merit of their research and the clarity of their presentation. Please join us in congratulating this year’s awardees. Nasima AkhterUniversity of Rochester I study the…
-

Tips for finding a scientific narrative
How many robotic talks and lectures have you experienced in your scientific journey? Probably more than you can count! As scientists, we typically prioritize accuracy when communicating our work but sometimes neglect to ensure the audience remains engaged. One way to hold your audience’s attention during presentations is to use a universal communication tool: storytelling. …