
With genomic data from hundreds of thousands of people accumulating, geneticists are now able to mine these data for very rare, but very informative genetic variants, including loss-of-function alleles. For example, across the enormous “reference set” of human exomes announced at the 2014 American Society for Human Genetics Meeting, on average there’s a variant every six bases. In the first of our reports from the ASHG meeting, Exome Aggregation Consortium (ExAC) lead analyst Monkol Lek (Massachusetts General Hospital/Broad Institute), has written a practical guide for geneticists looking to explore their-favorite-genes in the publicly-available exome data. Thanks to Monkol and Daniel MacArthur! If you’d like to write a guest post for Genes to Genomes, contact editor Cristy Gelling: cgelling@thegsajournals.org
We live in an amazing time to do human genetics. Over the last five years, thanks to impressive advances in DNA sequencing technology, the research community has collected sequencing data on genetic variation from over 200,000 samples. This provides us, for the first time, with the ability to study genetic variants at very low frequencies in the general population. However, in order to perform this research it’s critical that these genetic data be brought together and analyzed in the same way to ensure that the genetic changes that we find are real, and not the artifacts of differences in sequencing technology or analytical pipelines.
This goal is what drives the Exome Aggregation Consortium (ExAC), an international coalition of investigators with a focus on data from exome sequencing — an approach that allows us to focus variant discovery on the regions of the genome that encode proteins, known collectively as the exome. To date the Consortium has accumulated and jointly analyzed exome data from nearly 92,000 individuals, and has prepared a publicly accessible data set spanning 61,486 of these individuals for use as a global “reference set”. While the individuals in the reference set aren’t necessarily healthy — many have adult-onset diseases such as type 2 diabetes and schizophrenia — we have removed individuals with severe pediatric diseases, making this (we believe) a reasonable comparison data set for childhood-onset Mendelian diseases.
On October 20th at the American Society of Human Genetics (ASHG) conference we announced release 0.1 of the ExAC data set in two forms, as a browser and a downloadable raw data file. This was not just a massive data release but also a massive collaborative effort, which is detailed here. Four weeks after the release, the ExAC browser has received over 120,000 page views from over 17,000 unique users, and the raw data has been downloaded by over 150 organizations. The annotation tools ANNOVAR and ATAV have provided updates that have incorporated the ExAC data and the developers of Combined Annotation Dependent Depletion (CADD) have provided corresponding CADD scores. The commercial tools from GoldenHelix and GeneTalk have also incorporated the ExAC data. As the lead analyst on the project for over 2 years, I’ve been thrilled with the response it has received and the kind words and valuable feedback from the research community.
This practical guide, which uses two example genes FBN1 and MECP2 is aimed at general users and how they can access information using the ExAC Browser.
FBN1 Example
John Belmont commented on Nature News and Twitter that, within the ExAC dataset, the FBN1 gene associated with Marfan syndrome has 11 subjects with Loss of function (LoF) mutations. If these are true disease-causing variants then it fits roughly with the 1 in 5000 incidence of this disease.
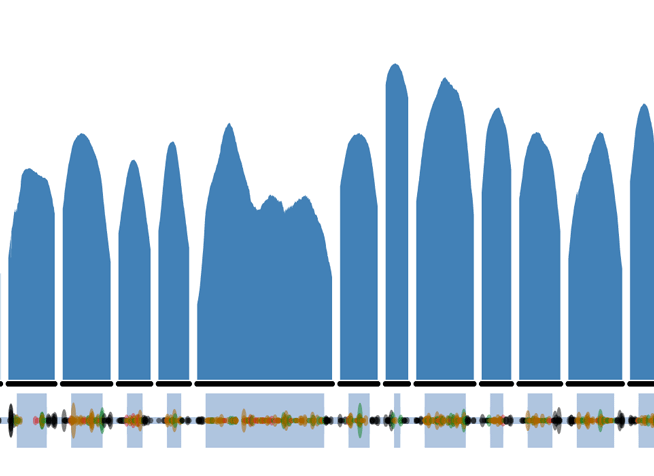
The FBN1 LoF variants can be directly viewed on the ExAC browser by searching FBN1 or clicking on this link and then clicking on the LoF button. Things to note on the FBN1 gene summary page:
- The coverage plot (affectionately called the Guilin plots ) is of the canonical transcript, using the Ensembl definition. This may not necessarily correspond to the clinically relevant transcript.
- The genomic coordinates uses GRCh37 and NOT the recently released GRCh38.
- Variant sites with multiple alleles are represented on separate rows.
- The functional annotation and corresponding protein consequence is from the most severe impact amongst the transcripts and may not affect all transcripts. As it is summarized from multiple transcripts, the amino acid position can sometimes appear out of order.
- Allele Number is the number of chromosomes, so is twice the number of individuals (maximum 2*61,486). Due to the nature of exome capture and quality thresholds applied, this will not always be at the maximum.
- All variant data displayed in the table can be downloaded as a CSV text file and opened in Excel to restore the columns and rows.
Amongst the 10 Loss of Function variants in FBN1
Using the stop-gained 15-48719948-G-C variant page is a good example to highlight important features:
- The histogram of Depth and Genotype Quality (GQ) is for individuals with the allele. Click on the “full site metrics” check box to display the histogram for all individuals with genotype calls (including those that are homozygous reference).
- The stop-gain variant does not affect all transcripts. The variant results in a missense change in the canonical transcript (ENST00000316623). In fact the canonical transcript has only 8/10 LoF mutations.
One of the upcoming features we are developing for the ExAC browser is the ability to view the sequencing reads from the reconstructed BAMs produced by the Genome Analysis Toolkit (GATK) Haplotype Caller using the –bamOutput option. For the splice acceptor variant 15-48760301-T-C, this is particularly useful to not only show the reads/bases supporting the SNP calls but also the reference sequence context and whether the acceptor site is canonical (i.e. ends in [T/C]AG).
Note: FBN1 is on the reverse strand

MECP2 Example
Loss of Function and ClinVar variants
Variants of the X-linked gene MECP2 can cause the neurodevelopmental disorder Rett syndrome, which affects mainly females. MECP2 LoF variants can be viewed by either following this link or searching MECP2 then clicking on the LoF button. In MECP2 there are 6 LoF variants. The stop-gained variant X-153296689-G-A has an allele count of 68, with 20 homozygous individuals. Currently the ExAC data set is not sex aware and does not differentiate between hemizygous males and homozygous females. An upcoming feature is to calculate these numbers correctly for variants on the X chromosome. The sex of each individual in ExAC was determined by heterozygosity on the X chromosome and normalized chromosome Y coverage.
Differentiating males and females from exome sequencing data, using chrX heterozygosity (X axis) and coverage on the Y chromosome (Y axis). Males form a cluster on the left, females on the bottom right. A small number of unassigned individuals are also visible, some of whom are probable Klinefelter cases.

Now for the stop-gained variant of interest, all 20 of the homozygous individuals are actually hemizygous males. Similar to the FBN1 example, the stop-gained annotation only affects 1/3 transcripts while the other two (including the canonical) have a missense (p.Thr197Met, p.Thr209Met) annotation. According to ClinVar, this variant is a missense variant and classified as benign. The LoF variants X-153296104-TCAGG-T and X-153296112-AGGTGGGG-A with homozygous individuals are also due to hemizygous males.
The variant X-153295997-C-T is an example of a pathogenic ClinVar variant in MECP2 that is claimed to be associated with neonatal severe encephalopathy in males. The 4 homozygous individuals in ExAC are actually 4 hemizygous males. It was later argued to be a rare variant rather than pathogenic but still remains classed as pathogenic in ClinVar!
Finally for a pathogenic variant in ClinVar not found on the ExAC browser, with genomic coordinates X-153296806. The coverage data also provided for download shows that this site has adequate coverage for variants to be detected.
tabix -h Panel.chrX.coverage.txt.gz X:153296806-153296806
Fraction of samples at various coverage.
| Chr | Pos | Mean | Median | 1x | 5x | 10x | 20x | 30x | 50x | >=100x |
|---|---|---|---|---|---|---|---|---|---|---|
| X | 153296806 | 72.81 | 72.00 | 1.0000 | 1.0000 | 0.9995 | 0.9918 | 0.9670 | 0.8025 | 0.3005 |
Looking more deeply at the insertion/deletions (indels) that result in frameshifts
Another advantage of having the ability to view sequencing reads is that users can now look at the reliability of the more difficult indels and other complex variant calls. The Exome Variant Server (EVS) is a fantastic resource for the research community but did not have features for researchers to scrutinize indel variant calls. This was particularly concerning for researchers when publishing on novel disease genes. In the case of a recently published paper on LMOD3, for instance, the presence of homozygous frameshift indels in EVS greatly concerned our collaborators; it was only through careful scrutiny of the raw data for these variants that we were able to reassure them that these were genotyping errors.
Firstly, let’s take a look at the 28 bp deletion X-153296090-CGGAGCTCTCGGGCTCAGGTGGAGGTGGG-C in MECP2 which results in a frameshift variant.

Now let’s see the reads for a 1 bp insertion X-153296070-A-AG from a heterozygous female.

Both of these frameshift mutations appear real and may cause intellectual disability, so why do they exist in a data set of individuals without severe diseases? I propose three possible reasons:
- There is an obvious drop in coverage where all the LoFs in MECP2 have accumulated. This may indicate a region difficult to capture or sequence and perhaps also challenging to detect variants.
- The shorter protein coding transcript ENST00000407218 avoids all but one LoF mutation (X-153296689-G-A) and may rescue some function lost in the larger isoforms.
- Lastly, the LoF mutations are towards the end of the gene and may result in a milder phenotype.
Investigating which of these possibilities may be contributing will require further detailed analysis. We welcome comments from MECP2 researchers regarding the LoF mutations in ExAC.
Tri and Quad allelic SNPs
Ending on an interesting point resulting from larger and larger data sets. The assumption that common variants remain bi-allelic is no longer valid, as with each new individual added there is a possibility of finding a new allele at a site where a bi-allelic variant is present. For example, the variant site rs2063690 is now a quad-allelic SNP – in other words, every possible base is present at this site in at least one individual in our data set! Furthermore, the figures below show three individuals who are heterozygous for the reference and each of the alternate alleles, while the last individual is heterozygous for two alternate alleles.
Heterozygous G/C (ref/alt)

Heterozygous G/A (ref/alt)

Heterozygous G/T (ref/alt)

Heterozygous C/A (alt/alt)

There is increasing urgency for the development of tools that deal appropriately with these multiallelic sites — approximately 7% of ExAC sites are now multi-allelic, and that fraction will grow as our sample size increases. That high rate of multiallelism shouldn’t be surprising, by the way; the ExAC dataset now (staggeringly) contains one variant every six bases on average, so it’s not a shock to see many cases where variant locations overlap.
Final thoughts
We’ve been gratified to see the rapid and positive response of the community to the ExAC data set. We still have plenty of work to do, though – and we’d love to get your feedback. If you have issues with the data set or the website, please drop us an email. For website bugs or feature requests you can also lodge a Github issue.
Many thanks to Daniel MacArthur for comments/feedback, writing introduction and final thoughts!
Guest posts are contributed by members of our community. The views expressed in guest posts are those of the author(s) and are not necessarily endorsed by the Genetics Society of America. If you'd like to write a guest post, e-mail communications@genetics-gsa.org.
View all posts by Guest Author »Read more in
-

Thank you, GSA community!
Thank you for being a member of the Genetics Society of America! As GSA’s current president, I am writing to tell you about Society projects and initiatives that we hope you will find useful in advancing your science and your career. Scientific research is a collaborative and exciting endeavor. Scientific societies like GSA exist to…
-

Where are they now? Rosalind Franklin Young Investigator Award recipients share updates on their research
Rosalind Franklin Young Investigator Award applications are open–make sure you submit your application or nomination of a colleague by September 30, 2024.
-

University of Minnesota researchers map genome of the last living wild horse species
The study, published in G3: Genes|Genomes|Genetics, is part of larger conservation efforts to save Przewalski’s horse.
-

Congratulations to the Spring 2024 DeLill Nasser Awardees!
GSA is pleased to announce the recipients of the DeLill Nasser Award for Professional Development in Genetics for Spring 2024! Given twice a year to graduate students and postdoctoral researchers, DeLill Nasser Awards support attendance at meetings and laboratory courses. The award is named in honor of DeLill Nasser, a long-time GSA supporter and National Science Foundation…
-

Carolyn Damilola: an NFS Rising Scientist on a lifelong quest to learn more
Carolyn Damilola is an NFS Rising Scientist from Nigeria doing respiratory system research and paving the way for scientists from underrepresented communities through mentorship.
-

What does a good microgrant proposal look like?
Members of the Microgrant Review Committee share their tips for a successful proposal.
-

The first piece of the facial recognition puzzle
New research in GENETICS gives a first peek at the molecular pathway involved in recognizing faces.
-

New Senior Editor Amy MacQueen joins GENETICS
A new senior editor is joining GENETICS in the Genome Integrity and Transmission section. We’re excited to welcome Amy MacQueen to the editorial team.
-

Block party on the zebrafish sex chromosome
Research in G3 identifies a gene regulatory block of the zebrafish genome responsible for overseeing the maternal-to-zygotic-transition.
-

Unraveling the mysteries of duckweed: epigenetic insights from Spirodela polyrhiza
Research published in G3 offers insight into the impact of DNA methylation on clonal propagation in asexually reproducing plants.
-

A microbiologist’s quest to understand CRISPR in bacterial self-defense
2024 Genetics Society of America Medal recipient Luciano Marraffini determined how CRISPR-Cas systems destroy genetic targets with precision, paving the way for gene editing technology development.
-

Unlocking mysteries of trait and disease heritability in dogs
2024 Edward Novitski Prize recipient Elaine Ostrander, a pioneer of the domestic dog model, discovered numerous genes affecting dog size, morphology, behavior, and disease susceptibility—many of which have relevance in humans.
-

GSA and collaborators Personal Genetics Education & Dialogue and Reclaiming STEM Institute launch NSF-funded BIO-LEAPS project to support culture change in genetics
We are thrilled to announce that the Genetics Society of America (GSA) is collaborating with the Personal Genetics Education & Dialogue (PGED) based in the Department of Genetics at Harvard Medical School, and the Reclaiming STEM Institute (RSI) on a Leading Culture Change Through Professional Societies of Biology (BIO-LEAPS) grant from the U.S. National Science…
-

Daman Saluja: Navigating Science and Policy in India
In the Paths to Science Policy series, we talk to individuals who have a passion for science policy and are active in advocacy through their various roles and careers. The series aims to inform and guide early career scientists interested in science policy. This series is brought to you by the GSA Early Career Scientist…
-

A fly geneticist’s journey into discovering rules of organ development
2024 George W. Beadle Award recipient Deborah Andrew discovered new genes and pathways in Drosophila salivary gland organogenesis. Now, her work can help optimize cell secretion in therapeutic applications and fight malaria.
-

Małgorzata Gazda: How receiving the DeLill Nasser Award helped her land her dream job
Have you ever experienced an event that changes the course of your life, or in this case, your career? Małgorzata (Gosia) Gazda is Assistant Professor at the University of Montreal and in 2022, she received the DeLill Nasser Award for Professional Development in Genetics, which she used to attend and present at the 2022 Population,…
-

Hongyu Zhao joins GENETICS as new Senior Editor
A new senior editor is joining GENETICS in the Statistical Genetics and Genomics section. We’re excited to welcome Hongyu Zhao to the editorial team.
-

GSA Member Julio Molina Pineda Receives DeLill Nasser Award, Shines at TAGC 2024
“At any career stage, the GSA membership is an amazing investment for any genetics professional!” Julio Molina Pineda is a PhD Candidate in Cell and Molecular Biology and a Research Assistant at the University of Arkansas, and a Doctoral Academy Fellow at the Lewis Lab. In 2023, Julio was awarded the DeLill Nasser Award for…
-

In Memoriam: Ellsworth Herman Grell (1932–2023), a pioneer of Drosophila genome engineering and annotation
Ellsworth (Ed) Grell blessed the Drosophila community through three enduring legacies: as a pioneer of chromosome mechanics, as a primary organizer and synthesizer of genetic knowledge in Drosophila, and as a graceful mentor to those fortunate to have known him personally. Ed grew up in rural Nebraska, completed his undergraduate studies at Iowa State, and…
-

Congratulations to the #Fungal24 Poster Award winners!
We are pleased to announce the recipients of the GSA Poster Awards for posters presented at the 32nd Fungal Genetics Conference! Undergraduate and graduate student members of GSA were eligible for the awards, and a hard-working team of judges made the determinations. Congratulations to all! Felicia Ebot Ojong, The University of Georgia My research is focused…
-

Poster presentation tips for TAGC 2024
You’ve been selected to present a poster at The Allied Genetics Conference 2024 in March—you’ve celebrated, made plans to attend, now what? This is an exciting opportunity to showcase your research and engage with fellow members of the genetics community, so you want to make sure you’re prepared. We wanted to offer you some tips…
-

Maximize your TAGC 2024 experience
A guide to all that National Harbor & DC have to offer Are you joining us for The Allied Genetics Conference 2024 in March? Make the most of your #TAGC24 experience in National Harbor! We know the science will keep you busy, but you deserve to unwind and have some fun, so we’ve curated a…
-

Early Career Leadership Spotlight: Sarah Petrosky
We’re taking time to get to know the members of the GSA’s Early Career Scientist Committees. Join us to learn more about our early career scientist advocates. Sarah PetroskyMultimedia SubcommitteeUniversity of Pittsburgh Research Interest I am interested in understanding adaptation that has been happening recently in populations by dissecting the ways that genes underlying an adaptation…
-

TAGC 2024 Early Career Award Winners
GSA is pleased to announce the winners of the early career awards presented at The Allied Genetics Conference 2024. These awards are specific to particular TAGC communities and recognize early career scientists’ outstanding work on their respective research organisms. The awardees will present their talks in keynote sessions at TAGC 2024. Don’t miss the opportunity…
-

Preeminent geneticists recognized with revamped GSA Awards
In 2022, GSA’s Board of Directors launched an audit to review the five major awards conferred by the Society. Today, we are thrilled to announce the recipients of the reimagined GSA Awards, including the new Genetics Society of America Early Career Medal. The scientists honored this year are recognized by their peers for their outstanding…
-

Fly Board funds outreach programs to spread the word about Drosophila research
In 2020, the Fly Board voted to use part of its reserve fund to support efforts to increase trainee participation as well as equity and diversity in the Drosophila community. An awards committee decides how the money will be spent each year, and from 2020–2022, the committee posted a very broad call for applications from…
-

New members of the GSA Board of Directors: 2024–2026
We are pleased to announce the election of four new leaders to the GSA Board of Directors: 2024 Vice President/2025 President Brenda Andrews Professor, University of Toronto It’s an honor to continue my association with the Society by serving as Vice President of the Board of Directors. I have broad knowledge of the ongoing activities…
-

Policy changes are closing the door on the “American Dream”
The American Dream once promised that talent and hard work could open doors. For early career scientists, those doors are now closing. Research is being paused or studies outright canceled, funding delayed or completely pulled, and careers cut short—not because of bad science, but because of changing political priorities. Researchers are being forced to choose: leave science or leave the country. These setbacks send a clear message: the U.S. commitment to supporting the next generation of scientists is waning.
-

Congratulations to the Worm 2025 GSA Poster Award Recipients
We are pleased to announce the GSA Poster Award recipients from the 25th International Worm Meeting! Undergraduate and graduate student GSA members were eligible for these awards, and a hard-working team of judges made the determinations. Congratulations to all! Kehinde Joan Abayomi, Mount Desert Island Biological Laboratory Category: Gene Regulation & Genomics Poster title: “Transcriptome…
-

GENETICS Expands Primers Section
Beth De Stasio of Lawrence University will serve as Senior Editor and work with a newly formed team of six Associate Editors to expand the publication of Primers in GENETICS.