
Guest post by Kathryn “Kaylee” Mueller, Phase Genomics.
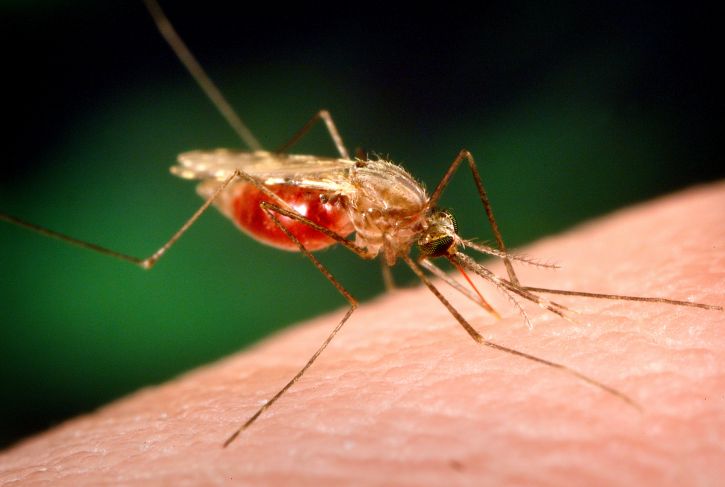
Plasmodium parasites—the microbes that cause malaria—are right at home in the tropics. After all, tropical regions harbor the two animals that the malaria parasites need to complete their complex lifecycle: female Anopheles mosquitoes and human beings. And in 2017 alone, Plasmodiumracked up 219 million cases of malaria, with 435,000 deaths.
Though malaria has spread over time across the world’s tropical areas, one region in particular—sub-Saharan Africa—unquestionably remains ground zero for the disease. African countries accounted for more than 90 percent of malaria cases and malaria deaths in 2017, according to the World Health Organization. Plasmodium‘s options for mosquito hosts in sub-Saharan Africa may explain malaria’s outsized presence there.
“Africa suffers disproportionately,” says Nora Besansky, a malaria researcher at the University of Notre Dame. “This is mainly due to the dominance of two highly efficient mosquito vectors of human malaria found throughout that region: Anopheles gambiae and Anopheles funestus.”
The twin scourges of malaria in Africa
Researchers long paid attention to An. gambiae due to its knack for transmitting the deadliest of the Plasmodium species. But scientists were slower to recognize that An. funestus, the mosquito that shares much of An. gambiae‘s range, also shows grim aptitude as a malaria vector. For example, An. funestus strongly prefers to bite humans over other animals. In addition, it tends to hang out indoors, gaining more opportunities to bite people and potentially spread malaria.
A game of catch-up
Yet scientists know much less about An. funestus than An. gambiae. Laboratory strains of An. funestus have been around for just 19 years, compared to more than 65 years for An. gambiae. The An. gambiae genome was released in 2002, the second for insects. The An. funestus genome came out in 2015, but its fragmented assembly consists of almost 1,400 scaffolds—despite a nuclear genome of just three chromosome pairs—resulting in fragmented genes, missing gene clusters, and little reliable structural information about the genome.
Assembling a better genome
Besansky is part of a team that decided to start over. They have produced a new, chromosome-scale assembly of the An. funestus genome—all from a pool of individuals of an An. funestus laboratory strain. The team used PacBio sequencing to produce long-read sequences, which they assembled into larger contigs and then filtered to remove duplicated sequences.
“Since this was a colony of mosquitos, there was high sequence duplication present in the data,” said Jay Ghurye, a member of the team and doctoral student at the University of Maryland, College Park. “We had to remove the duplications before scaffolding them using Hi-C data.”
That Hi-C data came from Phase Genomics, a partner in this project. Phase Genomics used its Proximo Hi-C approach to collapse the team’s contigs into longer scaffolds and—ultimately—produce three chromosome-length assemblies for An. funestus.
A complete roadmap
Based on contig and scaffold length alone, the new genome for An. funestusis a 100-fold improvement on the prior assembly, according to the researchers.
“Just as driving from New York to Los Angeles is facilitated by a roadmap, genetic research also is more powerful, efficient, and accurate if the 260 million puzzle pieces of nucleotides in the nuclear genome are properly ordered and oriented,” said Dr. Besansky.
The team hopes that this more complete picture of the An. funestus genome will help scientists understand what makes it such an effective malaria vector and come up with measures to countermand it.
One large to-do list for one little bug
Researchers already have a bevy of burning questions about An. funestus.
For instance, according to the World Health Organization, An. funestus is fast evolving resistance to the pyrethroid insecticides that are used to treat bed nets. The new genome includes features left out of the first assembly, such as genes arranged in tandem clusters. And at least one type of gene often found in these types of clusters—cytochrome P450 genes—has already been implicated in pyrethroid resistance in An. funestus.
Resistance is spreading in other ways too. Some An. funestus populations may have evolved behavioral adaptations to evade anti-mosquito controls. Scientists can now employ the new genome to help locate the genetic underpinnings of these traits.
With the new genome, researchers should also have an easier time detecting chromosomal rearrangements, such as inversions, among An. funestus populations.
“Inversions affect the spread of malaria, indirectly if not directly, because they typically contain hundreds or thousands of genes involved in climatic or local adaptation—allowing their mosquito carriers to fully exploit heterogeneous and otherwise challenging environments,” says Besansky.
An. funestus has obviously been busy, employing multiple approaches to dodge anti-mosquito measures. But all of these adaptations originate from the same source: the An. funestus genome. With that toolbox now completely sequenced, aligned, assembled and annotated, scientists may, at last, have the right blueprints to bring this malaria vector to heel.
Guest posts are contributed by members of our community. The views expressed in guest posts are those of the author(s) and are not necessarily endorsed by the Genetics Society of America. If you'd like to write a guest post, e-mail communications@genetics-gsa.org.
View all posts by Guest Author »Read more in
-

Thank you, GSA community!
Thank you for being a member of the Genetics Society of America! As GSA’s current president, I am writing to tell you about Society projects and initiatives that we hope you will find useful in advancing your science and your career. Scientific research is a collaborative and exciting endeavor. Scientific societies like GSA exist to…
-

Where are they now? Rosalind Franklin Young Investigator Award recipients share updates on their research
Rosalind Franklin Young Investigator Award applications are open–make sure you submit your application or nomination of a colleague by September 30, 2024.
-

University of Minnesota researchers map genome of the last living wild horse species
The study, published in G3: Genes|Genomes|Genetics, is part of larger conservation efforts to save Przewalski’s horse.
-

Congratulations to the Spring 2024 DeLill Nasser Awardees!
GSA is pleased to announce the recipients of the DeLill Nasser Award for Professional Development in Genetics for Spring 2024! Given twice a year to graduate students and postdoctoral researchers, DeLill Nasser Awards support attendance at meetings and laboratory courses. The award is named in honor of DeLill Nasser, a long-time GSA supporter and National Science Foundation…
-

Carolyn Damilola: an NFS Rising Scientist on a lifelong quest to learn more
Carolyn Damilola is an NFS Rising Scientist from Nigeria doing respiratory system research and paving the way for scientists from underrepresented communities through mentorship.
-

What does a good microgrant proposal look like?
Members of the Microgrant Review Committee share their tips for a successful proposal.
-

The first piece of the facial recognition puzzle
New research in GENETICS gives a first peek at the molecular pathway involved in recognizing faces.
-

New Senior Editor Amy MacQueen joins GENETICS
A new senior editor is joining GENETICS in the Genome Integrity and Transmission section. We’re excited to welcome Amy MacQueen to the editorial team.
-

Block party on the zebrafish sex chromosome
Research in G3 identifies a gene regulatory block of the zebrafish genome responsible for overseeing the maternal-to-zygotic-transition.
-

Unraveling the mysteries of duckweed: epigenetic insights from Spirodela polyrhiza
Research published in G3 offers insight into the impact of DNA methylation on clonal propagation in asexually reproducing plants.
-

A microbiologist’s quest to understand CRISPR in bacterial self-defense
2024 Genetics Society of America Medal recipient Luciano Marraffini determined how CRISPR-Cas systems destroy genetic targets with precision, paving the way for gene editing technology development.
-

Unlocking mysteries of trait and disease heritability in dogs
2024 Edward Novitski Prize recipient Elaine Ostrander, a pioneer of the domestic dog model, discovered numerous genes affecting dog size, morphology, behavior, and disease susceptibility—many of which have relevance in humans.
-

GSA and collaborators Personal Genetics Education & Dialogue and Reclaiming STEM Institute launch NSF-funded BIO-LEAPS project to support culture change in genetics
We are thrilled to announce that the Genetics Society of America (GSA) is collaborating with the Personal Genetics Education & Dialogue (PGED) based in the Department of Genetics at Harvard Medical School, and the Reclaiming STEM Institute (RSI) on a Leading Culture Change Through Professional Societies of Biology (BIO-LEAPS) grant from the U.S. National Science…
-

Daman Saluja: Navigating Science and Policy in India
In the Paths to Science Policy series, we talk to individuals who have a passion for science policy and are active in advocacy through their various roles and careers. The series aims to inform and guide early career scientists interested in science policy. This series is brought to you by the GSA Early Career Scientist…
-

A fly geneticist’s journey into discovering rules of organ development
2024 George W. Beadle Award recipient Deborah Andrew discovered new genes and pathways in Drosophila salivary gland organogenesis. Now, her work can help optimize cell secretion in therapeutic applications and fight malaria.
-

Małgorzata Gazda: How receiving the DeLill Nasser Award helped her land her dream job
Have you ever experienced an event that changes the course of your life, or in this case, your career? Małgorzata (Gosia) Gazda is Assistant Professor at the University of Montreal and in 2022, she received the DeLill Nasser Award for Professional Development in Genetics, which she used to attend and present at the 2022 Population,…
-

Hongyu Zhao joins GENETICS as new Senior Editor
A new senior editor is joining GENETICS in the Statistical Genetics and Genomics section. We’re excited to welcome Hongyu Zhao to the editorial team.
-

GSA Member Julio Molina Pineda Receives DeLill Nasser Award, Shines at TAGC 2024
“At any career stage, the GSA membership is an amazing investment for any genetics professional!” Julio Molina Pineda is a PhD Candidate in Cell and Molecular Biology and a Research Assistant at the University of Arkansas, and a Doctoral Academy Fellow at the Lewis Lab. In 2023, Julio was awarded the DeLill Nasser Award for…
-

In Memoriam: Ellsworth Herman Grell (1932–2023), a pioneer of Drosophila genome engineering and annotation
Ellsworth (Ed) Grell blessed the Drosophila community through three enduring legacies: as a pioneer of chromosome mechanics, as a primary organizer and synthesizer of genetic knowledge in Drosophila, and as a graceful mentor to those fortunate to have known him personally. Ed grew up in rural Nebraska, completed his undergraduate studies at Iowa State, and…
-

Congratulations to the #Fungal24 Poster Award winners!
We are pleased to announce the recipients of the GSA Poster Awards for posters presented at the 32nd Fungal Genetics Conference! Undergraduate and graduate student members of GSA were eligible for the awards, and a hard-working team of judges made the determinations. Congratulations to all! Felicia Ebot Ojong, The University of Georgia My research is focused…
-

Poster presentation tips for TAGC 2024
You’ve been selected to present a poster at The Allied Genetics Conference 2024 in March—you’ve celebrated, made plans to attend, now what? This is an exciting opportunity to showcase your research and engage with fellow members of the genetics community, so you want to make sure you’re prepared. We wanted to offer you some tips…
-

Maximize your TAGC 2024 experience
A guide to all that National Harbor & DC have to offer Are you joining us for The Allied Genetics Conference 2024 in March? Make the most of your #TAGC24 experience in National Harbor! We know the science will keep you busy, but you deserve to unwind and have some fun, so we’ve curated a…
-

Early Career Leadership Spotlight: Sarah Petrosky
We’re taking time to get to know the members of the GSA’s Early Career Scientist Committees. Join us to learn more about our early career scientist advocates. Sarah PetroskyMultimedia SubcommitteeUniversity of Pittsburgh Research Interest I am interested in understanding adaptation that has been happening recently in populations by dissecting the ways that genes underlying an adaptation…
-

TAGC 2024 Early Career Award Winners
GSA is pleased to announce the winners of the early career awards presented at The Allied Genetics Conference 2024. These awards are specific to particular TAGC communities and recognize early career scientists’ outstanding work on their respective research organisms. The awardees will present their talks in keynote sessions at TAGC 2024. Don’t miss the opportunity…
-

Preeminent geneticists recognized with revamped GSA Awards
In 2022, GSA’s Board of Directors launched an audit to review the five major awards conferred by the Society. Today, we are thrilled to announce the recipients of the reimagined GSA Awards, including the new Genetics Society of America Early Career Medal. The scientists honored this year are recognized by their peers for their outstanding…
-

Fly Board funds outreach programs to spread the word about Drosophila research
In 2020, the Fly Board voted to use part of its reserve fund to support efforts to increase trainee participation as well as equity and diversity in the Drosophila community. An awards committee decides how the money will be spent each year, and from 2020–2022, the committee posted a very broad call for applications from…
-

New members of the GSA Board of Directors: 2024–2026
We are pleased to announce the election of four new leaders to the GSA Board of Directors: 2024 Vice President/2025 President Brenda Andrews Professor, University of Toronto It’s an honor to continue my association with the Society by serving as Vice President of the Board of Directors. I have broad knowledge of the ongoing activities…
-

An automated analytical pipeline for solving the GWAS puzzle
A new bioinformatic GWAS pipeline enables automated, multi-faceted analysis of the
combinatorial effect of multiple variants on a genetic trait. -

A year in a Drosophila lab, measured in carbon
Research published in GENETICS breaks down the total carbon emissions from a PhD student’s research, giving insights into where sustainability efforts matter most.
-

Congratulations to the Fall 2025 DeLill Nasser Awardees!
We’re thrilled to announce the Fall 2025 recipients of the DeLill Nasser Award for Professional Development in Genetics! Awarded twice a year, these grants help graduate students and postdocs take the next step in their careers—whether that’s attending a scientific meeting, participating in a lab course, or connecting with the broader genetics community. The award honors…