
The December issue of GENETICS is out now! Check out the highlights below or the full Table of Contents here.
Ectopic centromere nucleation by CENP-A in fission yeast, pp. 1433–1446
Marlyn Gonzalez, Haijin He, Qianhua Dong, Siyu Sun, and Fei Li
The mechanisms protecting the cell against formation of ectopic centromeres (neocentromeres) are poorly understood. Gonzalez et al. show that overexpression of the centromere-specific histone H3 variant CENP-A in fission yeast results in the assembly of ectopic CENP-A chromatin with features of neocentromeres. Intriguingly, this assembly is suppressed by overexpression of histone H3 or H4. The N-terminal domain of CENP-A prevented ectopic CENP-A assembly via ubiquitin-dependent proteolysis.
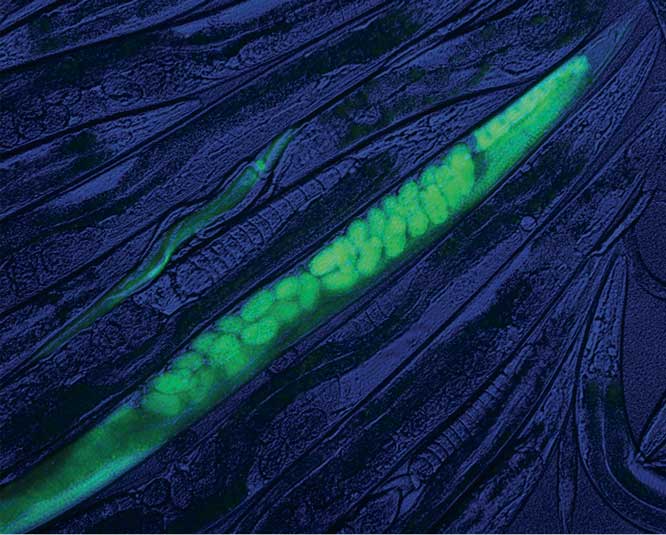
Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans, pp. 1347–1356
Alexandre Paix, Yuemeng Wang, Harold E. Smith, Chih-Yung S. Lee, Deepika Calidas, Tu Lu, Jarrett Smith, Helen Schmidt, Michael W. Krause, and Geraldine Seydoux
Paix et al. demonstrate that homology-dependent repair of Cas9-induced double-strand breaks is very robust in C. elegans and requires only short regions of homology. The practical implication is that it is possible to introduce edits of any size in the C. elegansgenome using oligonucleotides and PCR fragments with homology arms that are only 30-60 bases long.
The effects of demography and long-term selection on the accuracy of genomic prediction with sequence data, pp. 1671–1684
Iona M. MacLeod, Ben J. Hayes, and Michael E. Goddard
Dense SNP genotypes can be used to predict genetic or phenotypic outcomes in individuals for a complex trait, such as disease risk in humans and production traits in crops or livestock. MacLeod et al. evaluate the potential for improving the accuracy of genomic predictions by using sequence data compared to high density SNP arrays. They simulated data with contrasting population history, both with and without long-term purifying selection and tested two common analytical methods. The increase in accuracy achieved with sequence data varied considerably, demonstrating important dependencies and interactions with method of analysis, demographic history and long-term selection.
Transcriptome analysis indicates considerable divergence in alternative splicing between duplicated genes in Arabidopsis thaliana, pp. 1473–1481
David C. Tack, William R. Pitchers, and Keith L. Adams
Changes in alternative splicing of duplicated genes can generate new protein isoforms or alter gene expression. Tack et al. show that alternative splicing events have diverged considerably between duplicate genes in Arabidopsis. Most of the conserved splicing events showed divergence in event frequency, and most duplicated genes have diverged in alternative splicing-induced RNA decay.
A novel targeted learning method for quantitative trait loci mapping, pp. 1369–1376
Hui Wang, Zhongyang Zhang, Sherri Rose, and Mark van der Laan
Wang et al. present a semiparametric approach to QTL mapping that requires fewer restrictive assumptions about the data and accommodates machine learning algorithms with statistical inference. In simulations, the method was substantially less biased than other approaches, and it provided improved estimates and QTL rankings. When applied to a barley data set, the approach displayed less noise than composite interval mapping.
Bayesian inference of shared recombination hotspots between humans and chimpanzees, pp. 1621–1628
Ying Wang and Bruce Rannala
Several population genetic analyses have failed to detect recombination hotspots shared between humans and chimpanzees, despite their close genetic relationship. Using a Bayesian method and focusing on chromosome 21, Wang and Rannala analyzed chimp and human SNP data from recent resequencing projects. They also reanalyzed data from a previous study of human recombination hotspots in the MHC region. In both cases, the authors find strong evidence for shared hotspots between the two species.
The distribution of pairwise genetic distances: a tool for investigating disease transmission, pp. 1395–1404
Colin J. Worby, Hsiao-Han Chang, William P. Hanage, and Marc Lipsitch
Pathogen genome sequencing is an increasingly common tool in disease outbreak investigations, but the relationship between genetic distance and epidemiological factors is not well understood. Worby et al. investigated the distribution of genetic distance between isolates sampled during an outbreak, along with the effect of within-host dynamics and transmission bottleneck size. The authors derived an approximation to this distribution for testing transmission route hypotheses. This approach avoids the intensive computation of many existing methods but can identify a greater proportion of transmission routes.
Epigenetic control of learning and memory in Drosophila by Tip60 HAT action, pp.1571–1586
Songjun Xu, Rona Wilf, Trisha Menon, Priyalakshmi Panikker, Jessica Sarthi, and Felice Elefant
Xu et al. demonstrate that the histone acetyltransferase (HAT) Tip60 plays an epigenetic transcriptional regulatory role in cognitive function in Drosophila. Dysregulation of Tip60 HAT levels in the mushroom body causes memory deficits and misexpression of Tip60 cognition-linked targets. Remarkably, the learning and memory deficits induced by Alzheimer’s disease-associated amyloid precursor protein (APP) were rescued by increasing mushroom body Tip60 HAT levels. These results highlight the clinical potential of HAT activators.
Read more in
-

Thank you, GSA community!
Thank you for being a member of the Genetics Society of America! As GSA’s current president, I am writing to tell you about Society projects and initiatives that we hope you will find useful in advancing your science and your career. Scientific research is a collaborative and exciting endeavor. Scientific societies like GSA exist to…
-

Where are they now? Rosalind Franklin Young Investigator Award recipients share updates on their research
Rosalind Franklin Young Investigator Award applications are open–make sure you submit your application or nomination of a colleague by September 30, 2024.
-

University of Minnesota researchers map genome of the last living wild horse species
The study, published in G3: Genes|Genomes|Genetics, is part of larger conservation efforts to save Przewalski’s horse.
-

Congratulations to the Spring 2024 DeLill Nasser Awardees!
GSA is pleased to announce the recipients of the DeLill Nasser Award for Professional Development in Genetics for Spring 2024! Given twice a year to graduate students and postdoctoral researchers, DeLill Nasser Awards support attendance at meetings and laboratory courses. The award is named in honor of DeLill Nasser, a long-time GSA supporter and National Science Foundation…
-

Carolyn Damilola: an NFS Rising Scientist on a lifelong quest to learn more
Carolyn Damilola is an NFS Rising Scientist from Nigeria doing respiratory system research and paving the way for scientists from underrepresented communities through mentorship.
-

What does a good microgrant proposal look like?
Members of the Microgrant Review Committee share their tips for a successful proposal.
-

The first piece of the facial recognition puzzle
New research in GENETICS gives a first peek at the molecular pathway involved in recognizing faces.
-

New Senior Editor Amy MacQueen joins GENETICS
A new senior editor is joining GENETICS in the Genome Integrity and Transmission section. We’re excited to welcome Amy MacQueen to the editorial team.
-

Block party on the zebrafish sex chromosome
Research in G3 identifies a gene regulatory block of the zebrafish genome responsible for overseeing the maternal-to-zygotic-transition.
-

Unraveling the mysteries of duckweed: epigenetic insights from Spirodela polyrhiza
Research published in G3 offers insight into the impact of DNA methylation on clonal propagation in asexually reproducing plants.
-

A microbiologist’s quest to understand CRISPR in bacterial self-defense
2024 Genetics Society of America Medal recipient Luciano Marraffini determined how CRISPR-Cas systems destroy genetic targets with precision, paving the way for gene editing technology development.
-

Unlocking mysteries of trait and disease heritability in dogs
2024 Edward Novitski Prize recipient Elaine Ostrander, a pioneer of the domestic dog model, discovered numerous genes affecting dog size, morphology, behavior, and disease susceptibility—many of which have relevance in humans.
-

GSA and collaborators Personal Genetics Education & Dialogue and Reclaiming STEM Institute launch NSF-funded BIO-LEAPS project to support culture change in genetics
We are thrilled to announce that the Genetics Society of America (GSA) is collaborating with the Personal Genetics Education & Dialogue (PGED) based in the Department of Genetics at Harvard Medical School, and the Reclaiming STEM Institute (RSI) on a Leading Culture Change Through Professional Societies of Biology (BIO-LEAPS) grant from the U.S. National Science…
-

Daman Saluja: Navigating Science and Policy in India
In the Paths to Science Policy series, we talk to individuals who have a passion for science policy and are active in advocacy through their various roles and careers. The series aims to inform and guide early career scientists interested in science policy. This series is brought to you by the GSA Early Career Scientist…
-

A fly geneticist’s journey into discovering rules of organ development
2024 George W. Beadle Award recipient Deborah Andrew discovered new genes and pathways in Drosophila salivary gland organogenesis. Now, her work can help optimize cell secretion in therapeutic applications and fight malaria.
-

Małgorzata Gazda: How receiving the DeLill Nasser Award helped her land her dream job
Have you ever experienced an event that changes the course of your life, or in this case, your career? Małgorzata (Gosia) Gazda is Assistant Professor at the University of Montreal and in 2022, she received the DeLill Nasser Award for Professional Development in Genetics, which she used to attend and present at the 2022 Population,…
-

Hongyu Zhao joins GENETICS as new Senior Editor
A new senior editor is joining GENETICS in the Statistical Genetics and Genomics section. We’re excited to welcome Hongyu Zhao to the editorial team.
-

GSA Member Julio Molina Pineda Receives DeLill Nasser Award, Shines at TAGC 2024
“At any career stage, the GSA membership is an amazing investment for any genetics professional!” Julio Molina Pineda is a PhD Candidate in Cell and Molecular Biology and a Research Assistant at the University of Arkansas, and a Doctoral Academy Fellow at the Lewis Lab. In 2023, Julio was awarded the DeLill Nasser Award for…
-

In Memoriam: Ellsworth Herman Grell (1932–2023), a pioneer of Drosophila genome engineering and annotation
Ellsworth (Ed) Grell blessed the Drosophila community through three enduring legacies: as a pioneer of chromosome mechanics, as a primary organizer and synthesizer of genetic knowledge in Drosophila, and as a graceful mentor to those fortunate to have known him personally. Ed grew up in rural Nebraska, completed his undergraduate studies at Iowa State, and…
-

Congratulations to the #Fungal24 Poster Award winners!
We are pleased to announce the recipients of the GSA Poster Awards for posters presented at the 32nd Fungal Genetics Conference! Undergraduate and graduate student members of GSA were eligible for the awards, and a hard-working team of judges made the determinations. Congratulations to all! Felicia Ebot Ojong, The University of Georgia My research is focused…
-

Poster presentation tips for TAGC 2024
You’ve been selected to present a poster at The Allied Genetics Conference 2024 in March—you’ve celebrated, made plans to attend, now what? This is an exciting opportunity to showcase your research and engage with fellow members of the genetics community, so you want to make sure you’re prepared. We wanted to offer you some tips…
-

Maximize your TAGC 2024 experience
A guide to all that National Harbor & DC have to offer Are you joining us for The Allied Genetics Conference 2024 in March? Make the most of your #TAGC24 experience in National Harbor! We know the science will keep you busy, but you deserve to unwind and have some fun, so we’ve curated a…
-

Early Career Leadership Spotlight: Sarah Petrosky
We’re taking time to get to know the members of the GSA’s Early Career Scientist Committees. Join us to learn more about our early career scientist advocates. Sarah PetroskyMultimedia SubcommitteeUniversity of Pittsburgh Research Interest I am interested in understanding adaptation that has been happening recently in populations by dissecting the ways that genes underlying an adaptation…
-

TAGC 2024 Early Career Award Winners
GSA is pleased to announce the winners of the early career awards presented at The Allied Genetics Conference 2024. These awards are specific to particular TAGC communities and recognize early career scientists’ outstanding work on their respective research organisms. The awardees will present their talks in keynote sessions at TAGC 2024. Don’t miss the opportunity…
-

Preeminent geneticists recognized with revamped GSA Awards
In 2022, GSA’s Board of Directors launched an audit to review the five major awards conferred by the Society. Today, we are thrilled to announce the recipients of the reimagined GSA Awards, including the new Genetics Society of America Early Career Medal. The scientists honored this year are recognized by their peers for their outstanding…
-

Fly Board funds outreach programs to spread the word about Drosophila research
In 2020, the Fly Board voted to use part of its reserve fund to support efforts to increase trainee participation as well as equity and diversity in the Drosophila community. An awards committee decides how the money will be spent each year, and from 2020–2022, the committee posted a very broad call for applications from…
-

New members of the GSA Board of Directors: 2024–2026
We are pleased to announce the election of four new leaders to the GSA Board of Directors: 2024 Vice President/2025 President Brenda Andrews Professor, University of Toronto It’s an honor to continue my association with the Society by serving as Vice President of the Board of Directors. I have broad knowledge of the ongoing activities…
-

Congratulations to the 2025 DeLill Nasser Awardees!
We’re thrilled to announce the Spring 2025 recipients of the DeLill Nasser Award for Professional Development in Genetics! Awarded twice a year, these grants help graduate students and postdocs take the next step in their careers—whether that’s attending a scientific meeting, participating in a lab course, or connecting with the broader genetics community. The award…
-

New resources for our mid-career members
The Genetics Society of America continuously evaluates the needs of our community, including members from across career stages. The newly established Engagement and Professional Development Committee (EPDC)—comprised of early career scientists (ECS), mid-career and established faculty, and non-academic staff— will provide guidance regarding professional development programming to ensure GSA is offering timely and relevant career…
-

Mapping the natural history of yeast in a science outreach program
New research published in G3: Genes|Genomes|Genetics lays out a geographical sampling activity tailored for middle school students that helps discover genetic diversity in yeast populations residing in North American oaks.